Ethiopian Food and Drug Authority is a governmental organization mandated by Food and Medicine Administration Proclamation 1112/2019, that has been in effect since February 2019, to ensure a high level of protection of public health and safety. Public trust and confidence in medical devices, and in the administrative systems by which they are regulated, are based on the safety and performance of such products throughout their life cycle.
The Good Manufacturing Practice or Quality management System inspection is one of the conformity assessment procedures that the Authority conducts to ensure that the Products are being produced in a facility where the Quality management system is established, implemented and maintained. Part four, Article 20, Sub-article 4 of the proclamation (click here to access the document) of the proclamation 1112 states that- “Any medicine or medical device shall be registered if the manufacturer complies with Good Manufacturing Practices, dossiers are evaluated and found to fulfill safety, quality, and efficacy or effectiveness, and as appropriate fulfills laboratory quality test requirements”.
Though Quality management system compliance certification is mandatory for registering a medical device with EFDA, the Authority further requires regulatory inspection of the established QMS/GMP within the manufacturing facility. This is true especially for high risk (class III and IV) medical devices.
To ensure that the requirements are met, the Authority has developed and is implementing guidelines for Medical device Good Manufacturing Practices that contains detail regulatory requirements that a medical device manufacturer must fulfil to register its products with EFDA.
Regulatory documents related to medical device manufacturers onsite inspection
Proclamation 1112/2019
As detailed above, Part four, Article 20, Sub-article 4 of the proclamation states that- “Any medicine or medical device shall be registered if the manufacturer complies with Good Manufacturing Practices, dossiers are evaluated and found to fulfill safety, quality, and efficacy or effectiveness, and as appropriate fulfills laboratory quality test requirements”.
Service Fee Regulation 370/2015
This is a Regulation issued by the Council of Ministers for Rate of Service Fees for Food, Medicine, Health Professional and Health Institutions Registration and Licensing. This regulation was issued by the Council pursuant to Article 5 of the Definition- of Powers and Duties of the Executive Organs of the Federal Democratic Republic of Ethiopia Proclamation No. 916/2015, Article 17(1) of the Federal Government of Ethiopia Financial Administration Proclamation No. 648/2009 and Article 55(1) of the Food, Medicine, and Health Care Administration and Control Proclamation No. 661/2009 (the previous proclamation that gave mandate to the Authority, issued by the House of Peoples Representative).
The detail of fees related to GMP onsite inspection for both medicine and medical device is provided in the following table. The table is the screenshot of the regulation mentioned above.
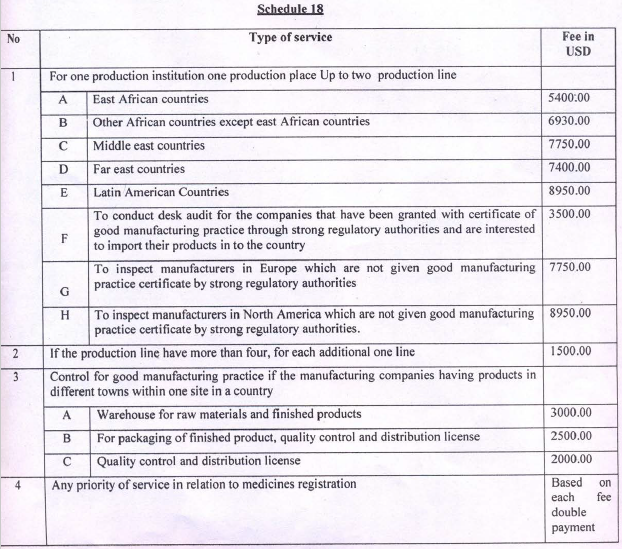
Though the payment rate increases depending on the manufacturing lines as per the regulation, the EFDA currently made product similarities and quantities the criteria to require manufacturers to pay the inspection service fees. This is linked with the number of days (and therefore expenses by the inspection team) it takes to complete the inspection. The categories of products and the associated service fee needed is always detailed in the official letter to be provided by the Authority.
Medical device Market Authorization Guidelines
There are three guidelines (General Market Authorization Guidelines, Non-In Vitro diagnostic Market Authorization guidelines and In Vitro diagnostic Market Authorization guidelines) specifically prepared for registration requirements of medical devices. All these guidelines require manufacturers to comply with GMP requirements.
In the General Guidelines for Medical devices Market Authorization, it is clearly stated that- all class III and IV medical devices require the manufacturing facility’s onsite inspection before registering the products. According to this guideline, the QMS for manufacturers of Class I & II devices and Class A & B IVD devices are normally not subjected to premarket on-site audit by EFDA. The exception is where assurance of sterility or of a measuring function is required, in which situation the associated procedures may be subject to the Authority’s premarket audit. The Authority can conduct an on-site audit of the manufacturing facilities of these Class I & II devices and Class A & B IVD devices if deemed necessary (e.g. Frequent lab test failures).
In general, even though EFDA has set procedures and requirements to assess the required GMP compliance of the manufacturers, the Authority currently conducts the onsite inspection of only three categories of devices- Condoms, Medical gloves including examination gloves and Rapid test kits. These products are selected due to their frequent post-market quality/performance failure complaints by users and the observations of the results from the Authority’s testing lab itself. It is expected that the EFDA will soon start enforcing the compliance of high-risk medical devices manufacturers with its regulatory GMP/QMS requirements.
What about the audited manufacturing facilities or their products approved by any of Stringent Regulatory Authorities (SRA)?
Stringent Regulatory Authority (SRA) is a competent government body or other entity that exercises a full legal right to control the use or sale of medical devices within its jurisdiction and may take enforcement action to ensure that medical devices marketed within its jurisdiction fully comply with legal requirements. An applicant claiming to have a Registration or Marketing Authorization certificate or GMP certificate issued by an SRA should provide supporting documents for its claim and if confirmed appropriate, the desk-review of the GMP compliance will be conducted by EFDA’s team of inspectors without travelling to the site. This will also decrease the manufacturer’s GMP assessment service fee payment to less than half (currently $3,500, as it can be seen from the table above).
At present, EFDA accepts the registration submissions (and therefore GMP compliance assessment) as an SRA or prequalified products application if they’re supported by registration/prequalification evidences from one or more of these Agencies or Organizations- The US Food and Drug Administration, Ministry of Health, Labour and Welfare (Japan), Therapeutic Goods Administration (Australia), EU Member States, MHRA (UK), Health Science Authority (Singapore), Health Canada and World Health organization (WHO).
Please do not hesitate to contact us if you have further questions or need additional guidance for registering your products with EFDA.